How to Design Pcr Primers Using Serial Cloner

Diagram of molecular cloning using bacteria and plasmids.
Molecular cloning is a set of experimental methods in molecular biology that are used to assemble recombinant DNA molecules and to direct their replication within host organisms.[1] The use of the word cloning refers to the fact that the method involves the replication of one molecule to produce a population of cells with identical DNA molecules. Molecular cloning generally uses DNA sequences from two different organisms: the species that is the source of the DNA to be cloned, and the species that will serve as the living host for replication of the recombinant DNA. Molecular cloning methods are central to many contemporary areas of modern biology and medicine.[2]
In a conventional molecular cloning experiment, the DNA to be cloned is obtained from an organism of interest, then treated with enzymes in the test tube to generate smaller DNA fragments. Subsequently, these fragments are then combined with vector DNA to generate recombinant DNA molecules. The recombinant DNA is then introduced into a host organism (typically an easy-to-grow, benign, laboratory strain of E. coli bacteria). This will generate a population of organisms in which recombinant DNA molecules are replicated along with the host DNA. Because they contain foreign DNA fragments, these are transgenic or genetically modified microorganisms (GMO).[3] This process takes advantage of the fact that a single bacterial cell can be induced to take up and replicate a single recombinant DNA molecule. This single cell can then be expanded exponentially to generate a large amount of bacteria, each of which contain copies of the original recombinant molecule. Thus, both the resulting bacterial population, and the recombinant DNA molecule, are commonly referred to as "clones". Strictly speaking, recombinant DNA refers to DNA molecules, while molecular cloning refers to the experimental methods used to assemble them. The idea arose that different DNA sequences could be inserted into a plasmid and that these foreign sequences would be carried into bacteria and digested as part of the plasmid. That is, these plasmids could serve as cloning vectors to carry genes.[4]
Virtually any DNA sequence can be cloned and amplified, but there are some factors that might limit the success of the process. Examples of the DNA sequences that are difficult to clone are inverted repeats, origins of replication, centromeres and telomeres. There is also a lower chance of success when inserting large-sized DNA sequences. Inserts larger than 10kbp have very limited success, but bacteriophages such as bacteriophage λ can be modified to successfully insert a sequence up to 40 kbp.[5]
History [edit]
Prior to the 1970s, the understanding of genetics and molecular biology was severely hampered by an inability to isolate and study individual genes from complex organisms. This changed dramatically with the advent of molecular cloning methods. Microbiologists, seeking to understand the molecular mechanisms through which bacteria restricted the growth of bacteriophage, isolated restriction endonucleases, enzymes that could cleave DNA molecules only when specific DNA sequences were encountered.[6] They showed that restriction enzymes cleaved chromosome-length DNA molecules at specific locations, and that specific sections of the larger molecule could be purified by size fractionation. Using a second enzyme, DNA ligase, fragments generated by restriction enzymes could be joined in new combinations, termed recombinant DNA. By recombining DNA segments of interest with vector DNA, such as bacteriophage or plasmids, which naturally replicate inside bacteria, large quantities of purified recombinant DNA molecules could be produced in bacterial cultures. The first recombinant DNA molecules were generated and studied in 1972.[7] [8]
Overview [edit]
Molecular cloning takes advantage of the fact that the chemical structure of DNA is fundamentally the same in all living organisms. Therefore, if any segment of DNA from any organism is inserted into a DNA segment containing the molecular sequences required for DNA replication, and the resulting recombinant DNA is introduced into the organism from which the replication sequences were obtained, then the foreign DNA will be replicated along with the host cell's DNA in the transgenic organism.
Molecular cloning is similar to polymerase chain reaction (PCR) in that it permits the replication of DNA sequence. The fundamental difference between the two methods is that molecular cloning involves replication of the DNA in a living microorganism, while PCR replicates DNA in an in vitro solution, free of living cells.
In silico cloning and simulations [edit]
Before actual cloning experiments are performed in the lab, most cloning experiments are planned in a computer, using specialized software. Although the detailed planning of the cloning can be done in any text editor, together with online utilities for e.g. PCR primer design, dedicated software exist for the purpose. Software for the purpose include for example ApE [1] (open source), DNAStrider [2] (open source), Serial Cloner [3] (gratis), Collagene [4] (open source), and SnapGene (commercial). These programs allow to simulate PCR reactions, restriction digests, ligations, etc., that is, all the steps described below.
Steps [edit]

The overall goal of molecular cloning is to take a gene of interest from one plasmid and insert it into another plasmid[9] This is done by performing PCR, digestive reaction, ligation reaction, and transformation.
In standard molecular cloning experiments, the cloning of any DNA fragment essentially involves seven steps: (1) Choice of host organism and cloning vector, (2) Preparation of vector DNA, (3) Preparation of DNA to be cloned, (4) Creation of recombinant DNA, (5) Introduction of recombinant DNA into host organism, (6) Selection of organisms containing recombinant DNA, (7) Screening for clones with desired DNA inserts and biological properties.
Notably, the growing capacity and fidelity of DNA synthesis platforms allows for increasingly intricate designs in molecular engineering. These projects may include very long strands of novel DNA sequence and/or test entire libraries simultaneously, as opposed to of individual sequences. These shifts introduce complexity that require design to move away from the flat nucleotide-based representation and towards a higher level of abstraction. Examples of such tools are GenoCAD, Teselagen [5] (free for academia) or GeneticConstructor [6] (free for academics).
Choice of host organism and cloning vector [edit]

Although a very large number of host organisms and molecular cloning vectors are in use, the great majority of molecular cloning experiments begin with a laboratory strain of the bacterium E. coli (Escherichia coli) and a plasmid cloning vector. E. coli and plasmid vectors are in common use because they are technically sophisticated, versatile, widely available, and offer rapid growth of recombinant organisms with minimal equipment.[3] If the DNA to be cloned is exceptionally large (hundreds of thousands to millions of base pairs), then a bacterial artificial chromosome[10] or yeast artificial chromosome vector is often chosen.
Specialized applications may call for specialized host-vector systems. For example, if the experimentalists wish to harvest a particular protein from the recombinant organism, then an expression vector is chosen that contains appropriate signals for transcription and translation in the desired host organism. Alternatively, if replication of the DNA in different species is desired (for example, transfer of DNA from bacteria to plants), then a multiple host range vector (also termed shuttle vector) may be selected. In practice, however, specialized molecular cloning experiments usually begin with cloning into a bacterial plasmid, followed by subcloning into a specialized vector.
Whatever combination of host and vector are used, the vector almost always contains four DNA segments that are critically important to its function and experimental utility:[3]
- DNA replication origin is necessary for the vector (and its linked recombinant sequences) to replicate inside the host organism
- one or more unique restriction endonuclease recognition sites to serves as sites where foreign DNA may be introduced
- a selectable genetic marker gene that can be used to enable the survival of cells that have taken up vector sequences
- a tag gene that can be used to screen for cells containing the foreign DNA

Cleavage of a DNA sequence containing the BamHI restriction site. The DNA is cleaved at the palindromic sequence to produce 'sticky ends'.
Preparation of vector DNA [edit]
The cloning vector is treated with a restriction endonuclease to cleave the DNA at the site where foreign DNA will be inserted. The restriction enzyme is chosen to generate a configuration at the cleavage site that is compatible with the ends of the foreign DNA (see DNA end). Typically, this is done by cleaving the vector DNA and foreign DNA with the same restriction enzyme, for example EcoRI. Most modern vectors contain a variety of convenient cleavage sites that are unique within the vector molecule (so that the vector can only be cleaved at a single site) and are located within a gene (frequently beta-galactosidase) whose inactivation can be used to distinguish recombinant from non-recombinant organisms at a later step in the process. To improve the ratio of recombinant to non-recombinant organisms, the cleaved vector may be treated with an enzyme (alkaline phosphatase) that dephosphorylates the vector ends. Vector molecules with dephosphorylated ends are unable to replicate, and replication can only be restored if foreign DNA is integrated into the cleavage site.[11]
Preparation of DNA to be cloned [edit]
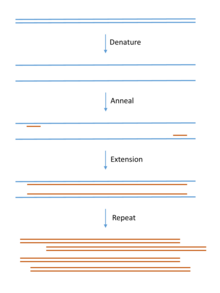
DNA for cloning is most commonly produced using PCR. Template DNA is mixed with bases (the building blocks of DNA), primers (short pieces of complementary single stranded DNA) and a DNA polymerase enzyme that builds the DNA chain. The mix goes through cycles of heating and cooling to produce large quantities of copied DNA.
For cloning of genomic DNA, the DNA to be cloned is extracted from the organism of interest. Virtually any tissue source can be used (even tissues from extinct animals),[12] as long as the DNA is not extensively degraded. The DNA is then purified using simple methods to remove contaminating proteins (extraction with phenol), RNA (ribonuclease) and smaller molecules (precipitation and/or chromatography). Polymerase chain reaction (PCR) methods are often used for amplification of specific DNA or RNA (RT-PCR) sequences prior to molecular cloning.
DNA for cloning experiments may also be obtained from RNA using reverse transcriptase (complementary DNA or cDNA cloning), or in the form of synthetic DNA (artificial gene synthesis). cDNA cloning is usually used to obtain clones representative of the mRNA population of the cells of interest, while synthetic DNA is used to obtain any precise sequence defined by the designer. Such a designed sequence may be required when moving genes across genetic codes (for example, from the mitochrondria to the nucleus)[13] or simply for increasing expression via codon optimization.[14]
The purified DNA is then treated with a restriction enzyme to generate fragments with ends capable of being linked to those of the vector. If necessary, short double-stranded segments of DNA (linkers) containing desired restriction sites may be added to create end structures that are compatible with the vector.[3] [11]
Creation of recombinant DNA with DNA ligase [edit]
The creation of recombinant DNA is in many ways the simplest step of the molecular cloning process. DNA prepared from the vector and foreign source are simply mixed together at appropriate concentrations and exposed to an enzyme (DNA ligase) that covalently links the ends together. This joining reaction is often termed ligation. The resulting DNA mixture containing randomly joined ends is then ready for introduction into the host organism.
DNA ligase only recognizes and acts on the ends of linear DNA molecules, usually resulting in a complex mixture of DNA molecules with randomly joined ends. The desired products (vector DNA covalently linked to foreign DNA) will be present, but other sequences (e.g. foreign DNA linked to itself, vector DNA linked to itself and higher-order combinations of vector and foreign DNA) are also usually present. This complex mixture is sorted out in subsequent steps of the cloning process, after the DNA mixture is introduced into cells.[3] [11]
Introduction of recombinant DNA into host organism [edit]
The DNA mixture, previously manipulated in vitro, is moved back into a living cell, referred to as the host organism. The methods used to get DNA into cells are varied, and the name applied to this step in the molecular cloning process will often depend upon the experimental method that is chosen (e.g. transformation, transduction, transfection, electroporation).[3] [11]
When microorganisms are able to take up and replicate DNA from their local environment, the process is termed transformation, and cells that are in a physiological state such that they can take up DNA are said to be competent.[15] In mammalian cell culture, the analogous process of introducing DNA into cells is commonly termed transfection. Both transformation and transfection usually require preparation of the cells through a special growth regime and chemical treatment process that will vary with the specific species and cell types that are used.
Electroporation uses high voltage electrical pulses to translocate DNA across the cell membrane (and cell wall, if present).[16] In contrast, transduction involves the packaging of DNA into virus-derived particles, and using these virus-like particles to introduce the encapsulated DNA into the cell through a process resembling viral infection. Although electroporation and transduction are highly specialized methods, they may be the most efficient methods to move DNA into cells.
Selection of organisms containing vector sequences [edit]
Whichever method is used, the introduction of recombinant DNA into the chosen host organism is usually a low efficiency process; that is, only a small fraction of the cells will actually take up DNA. Experimental scientists deal with this issue through a step of artificial genetic selection, in which cells that have not taken up DNA are selectively killed, and only those cells that can actively replicate DNA containing the selectable marker gene encoded by the vector are able to survive.[3] [11]
When bacterial cells are used as host organisms, the selectable marker is usually a gene that confers resistance to an antibiotic that would otherwise kill the cells, typically ampicillin. Cells harboring the plasmid will survive when exposed to the antibiotic, while those that have failed to take up plasmid sequences will die. When mammalian cells (e.g. human or mouse cells) are used, a similar strategy is used, except that the marker gene (in this case typically encoded as part of the kanMX cassette) confers resistance to the antibiotic Geneticin.
Screening for clones with desired DNA inserts and biological properties [edit]
Modern bacterial cloning vectors (e.g. pUC19 and later derivatives including the pGEM vectors) use the blue-white screening system to distinguish colonies (clones) of transgenic cells from those that contain the parental vector (i.e. vector DNA with no recombinant sequence inserted). In these vectors, foreign DNA is inserted into a sequence that encodes an essential part of beta-galactosidase, an enzyme whose activity results in formation of a blue-colored colony on the culture medium that is used for this work. Insertion of the foreign DNA into the beta-galactosidase coding sequence disables the function of the enzyme so that colonies containing transformed DNA remain colorless (white). Therefore, experimentalists are easily able to identify and conduct further studies on transgenic bacterial clones, while ignoring those that do not contain recombinant DNA.
The total population of individual clones obtained in a molecular cloning experiment is often termed a DNA library. Libraries may be highly complex (as when cloning complete genomic DNA from an organism) or relatively simple (as when moving a previously cloned DNA fragment into a different plasmid), but it is almost always necessary to examine a number of different clones to be sure that the desired DNA construct is obtained. This may be accomplished through a very wide range of experimental methods, including the use of nucleic acid hybridizations, antibody probes, polymerase chain reaction, restriction fragment analysis and/or DNA sequencing.[3] [11]
Applications [edit]
Molecular cloning provides scientists with an essentially unlimited quantity of any individual DNA segments derived from any genome. This material can be used for a wide range of purposes, including those in both basic and applied biological science. A few of the more important applications are summarized here.
Genome organization and gene expression [edit]
Molecular cloning has led directly to the elucidation of the complete DNA sequence of the genomes of a very large number of species and to an exploration of genetic diversity within individual species, work that has been done mostly by determining the DNA sequence of large numbers of randomly cloned fragments of the genome, and assembling the overlapping sequences.
At the level of individual genes, molecular clones are used to generate probes that are used for examining how genes are expressed, and how that expression is related to other processes in biology, including the metabolic environment, extracellular signals, development, learning, senescence and cell death. Cloned genes can also provide tools to examine the biological function and importance of individual genes, by allowing investigators to inactivate the genes, or make more subtle mutations using regional mutagenesis or site-directed mutagenesis. Genes cloned into expression vectors for functional cloning provide a means to screen for genes on the basis of the expressed protein's function.
Production of recombinant proteins [edit]
Obtaining the molecular clone of a gene can lead to the development of organisms that produce the protein product of the cloned genes, termed a recombinant protein. In practice, it is frequently more difficult to develop an organism that produces an active form of the recombinant protein in desirable quantities than it is to clone the gene. This is because the molecular signals for gene expression are complex and variable, and because protein folding, stability and transport can be very challenging.
Many useful proteins are currently available as recombinant products. These include--(1) medically useful proteins whose administration can correct a defective or poorly expressed gene (e.g. recombinant factor VIII, a blood-clotting factor deficient in some forms of hemophilia,[17] and recombinant insulin, used to treat some forms of diabetes[18]), (2) proteins that can be administered to assist in a life-threatening emergency (e.g. tissue plasminogen activator, used to treat strokes[19]), (3) recombinant subunit vaccines, in which a purified protein can be used to immunize patients against infectious diseases, without exposing them to the infectious agent itself (e.g. hepatitis B vaccine[20]), and (4) recombinant proteins as standard material for diagnostic laboratory tests.
Transgenic organisms [edit]
Once characterized and manipulated to provide signals for appropriate expression, cloned genes may be inserted into organisms, generating transgenic organisms, also termed genetically modified organisms (GMOs). Although most GMOs are generated for purposes of basic biological research (see for example, transgenic mouse), a number of GMOs have been developed for commercial use, ranging from animals and plants that produce pharmaceuticals or other compounds (pharming), herbicide-resistant crop plants, and fluorescent tropical fish (GloFish) for home entertainment.[1]
Gene therapy [edit]
Gene therapy involves supplying a functional gene to cells lacking that function, with the aim of correcting a genetic disorder or acquired disease. Gene therapy can be broadly divided into two categories. The first is alteration of germ cells, that is, sperm or eggs, which results in a permanent genetic change for the whole organism and subsequent generations. This "germ line gene therapy" is considered by many to be unethical in human beings.[21] The second type of gene therapy, "somatic cell gene therapy", is analogous to an organ transplant. In this case, one or more specific tissues are targeted by direct treatment or by removal of the tissue, addition of the therapeutic gene or genes in the laboratory, and return of the treated cells to the patient. Clinical trials of somatic cell gene therapy began in the late 1990s, mostly for the treatment of cancers and blood, liver, and lung disorders.[22]
Despite a great deal of publicity and promises, the history of human gene therapy has been characterized by relatively limited success.[22] The effect of introducing a gene into cells often promotes only partial and/or transient relief from the symptoms of the disease being treated. Some gene therapy trial patients have suffered adverse consequences of the treatment itself, including deaths. In some cases, the adverse effects result from disruption of essential genes within the patient's genome by insertional inactivation. In others, viral vectors used for gene therapy have been contaminated with infectious virus. Nevertheless, gene therapy is still held to be a promising future area of medicine, and is an area where there is a significant level of research and development activity.
References [edit]
- ^ a b Watson JD (2007). Recombinant DNA: genes and genomes: a short course. San Francisco: W.H. Freeman. ISBN978-0-7167-2866-5.
- ^ Patten CL, Glick BR, Pasternak J (2009). Molecular Biotechnology: Principles and Applications of Recombinant DNA. Washington, D.C: ASM Press. ISBN978-1-55581-498-4.
- ^ a b c d e f g h Brown T (2006). Gene cloning and DNA analysis: an introduction. Cambridge, MA: Blackwell Pub. ISBN978-1-4051-1121-8.
- ^ M., Grisham, Charles (2013-01-01). Biochemistry. Brooks/Cole, Cengage Learning. ISBN978-1133106296. OCLC 777722371.
- ^ Garret, Grisham (2010). Biochemistry. Belmont, CA, Brooks/Cole: Cengage Learning. p. 380.
- ^ Nathans D, Smith HO (1975). "Restriction endonucleases in the analysis and restructuring of dna molecules". Annual Review of Biochemistry. 44: 273–93. doi:10.1146/annurev.bi.44.070175.001421. PMID 166604.
- ^ Cohen SN, Chang AC, Boyer HW, Helling RB (Nov 1973). "Construction of biologically functional bacterial plasmids in vitro". Proceedings of the National Academy of Sciences of the United States of America. 70 (11): 3240–4. Bibcode:1973PNAS...70.3240C. doi:10.1073/pnas.70.11.3240. PMC427208. PMID 4594039.
- ^ Jackson DA, Symons RH, Berg P (Oct 1972). "Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli". Proceedings of the National Academy of Sciences of the United States of America. 69 (10): 2904–9. Bibcode:1972PNAS...69.2904J. doi:10.1073/pnas.69.10.2904. PMC389671. PMID 4342968.
- ^ "plasmid / plasmids | Learn Science at Scitable". www.nature.com . Retrieved 2017-12-06 .
- ^ Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M (Sep 1992). "Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector". Proceedings of the National Academy of Sciences of the United States of America. 89 (18): 8794–7. Bibcode:1992PNAS...89.8794S. doi:10.1073/pnas.89.18.8794. PMC50007. PMID 1528894.
- ^ a b c d e f Russell DW, Sambrook J (2001). Molecular cloning: a laboratory manual . Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory. ISBN978-0-87969-576-7.
- ^ Higuchi R, Bowman B, Freiberger M, Ryder OA, Wilson AC (1984). "DNA sequences from the quagga, an extinct member of the horse family". Nature. 312 (5991): 282–4. Bibcode:1984Natur.312..282H. doi:10.1038/312282a0. PMID 6504142. S2CID 4313241.
- ^ Boominathan, A; Vanhoozer, S; Basisty, N; Powers, K; Crampton, AL; Wang, X; Friedricks, N; Schilling, B; Brand, MD; O'Connor, MS (2 November 2016). "Stable nuclear expression of ATP8 and ATP6 genes rescues a mtDNA Complex V null mutant". Nucleic Acids Research. 44 (19): 9342–9357. doi:10.1093/nar/gkw756. PMC5100594. PMID 27596602.
- ^ Plotkin, J. B.; Kudla, G (2011). "Synonymous but not the same: The causes and consequences of codon bias". Nature Reviews Genetics. 12 (1): 32–42. doi:10.1038/nrg2899. PMC3074964. PMID 21102527.
- ^ Lederberg J (Feb 1994). "The transformation of genetics by DNA: an anniversary celebration of Avery, MacLeod and McCarty (1944)". Genetics. 136 (2): 423–6. doi:10.1093/genetics/136.2.423. PMC1205797. PMID 8150273.
- ^ Wirth R, Friesenegger A, Fiedler S (Mar 1989). "Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation". Molecular & General Genetics. 216 (1): 175–7. doi:10.1007/BF00332248. PMID 2659971. S2CID 25214157.
- ^ Oldenburg J, Dolan G, Lemm G (Jan 2009). "Haemophilia care then, now and in the future". Haemophilia. 15 Suppl 1: 2–7. doi:10.1111/j.1365-2516.2008.01946.x. PMID 19125934. S2CID 29118026.
- ^ The MJ (Nov 1989). "Human insulin: DNA technology's first drug". American Journal of Hospital Pharmacy. 46 (11 Suppl 2): S9-11. PMID 2690608.
- ^ Lewandowski C, Barsan W (Feb 2001). "Treatment of acute ischemic stroke". Annals of Emergency Medicine. 37 (2): 202–16. doi:10.1067/mem.2001.111573. PMID 11174240.
- ^ Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, Kong MS, Liang DC, Shau WY, Chen DS (Jun 1997). "Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group". The New England Journal of Medicine. 336 (26): 1855–9. doi:10.1056/NEJM199706263362602. PMID 9197213.
- ^ August JT (1997). Gene Therapy. 40. Academic Press. p. 508. ISBN978-0-08-058132-3.
- ^ a b Pfeifer A, Verma IM (2001). "Gene therapy: promises and problems". Annual Review of Genomics and Human Genetics. 2: 177–211. doi:10.1146/annurev.genom.2.1.177. PMID 11701648.
Further reading [edit]
- Matsumura, Ichiro (September 2015). "Why Johnny can't clone: Common pitfalls and not so common solutions". BioTechniques. 53 (3): IV–XIII. doi:10.2144/000114324. PMID 26345511. Archived from the original on 2015-09-16. Retrieved 2 February 2016.
External links [edit]
-
 Media related to Molecular cloning at Wikimedia Commons
Media related to Molecular cloning at Wikimedia Commons
How to Design Pcr Primers Using Serial Cloner
Source: https://en.wikipedia.org/wiki/Molecular_cloning
0 Response to "How to Design Pcr Primers Using Serial Cloner"
Post a Comment